
Losing, Yet Winning, in Life’s Genetic Lottery
An inherited mutation inspires a family-run patient advocacy group
Chapter 1
At 8 a.m. on Monday, August 27, 2007, a man in cycling attire rode a bicycle across the stage of a high school auditorium, circled back to the middle, stopped, then raised his hands in comic victory. He was greeted by loud and appreciative applause from his colleagues and friends, teachers at Stevens Point Area Senior High, where he had taught math for thirty-three years and directed many successful musicals. Greg Chelcun, fifty-six years old, was officially starting his retirement. As he had for the past ten years, Greg was clowning around on stage to ring in the new school year and to make a plug for the Stevens Point Area Education Association Scholarship fund, which he had founded to support post-secondary education for the school’s graduates. This year, however, after his scholarship plug, Greg didn’t join his colleagues in their seats; instead, he rode off the stage toward what he believed would be his golden years.

Greg and Cindy Chelcun
Twelve months later, when he returned to give a similar speech, Greg left his bike at home, along with his cycling jersey and shorts. He walked across the stage to the microphone, wearing a black fanny pack, its compartment swiveled to the front, visible to the hushed audience. Their eyes honed in on it. As if nothing was different, he encouraged his former colleagues to contribute to the scholarship fund. But this year, he finished his speech by explaining what everyone wanted to know: Greg had been diagnosed with late stage metastatic stomach cancer; the fanny pack contained a delivery pump for his daily dose of chemotherapy, which entered his body through a Medi-port implanted in his chest.
Stomach cancer is known as a “silent disease,” with virtually no detectable symptoms in the early stages when chemotherapy or surgery could actually make a difference. Six months later—eighteen months after retiring from the job he loved—Greg was dead.
Chapter 2
Just after his retirement, Greg had begun feeling full after very small meals. An upper endoscopy did not reveal any significant abnormalities, and biopsies were negative for cancer. Doctors tried removing his gall bladder. Greg’s symptoms worsened, however, leading to more tests, doctor visits, and finally emergency surgery to remove a tumor that had metastasized to his colon. He was eventually diagnosed with “cancer of unknown origin.” Despite all the technologies of modern medicine, doctors couldn’t locate where the cancer had started—was it in Greg’s stomach, pancreas, or appendix? Finally, after a second endoscopy, they pinpointed the cancer’s origin: his stomach. By then, it was Stage IV metastatic, meaning it had begun to spread. There was little the doctors could do.
Greg’s mother, Elaine, had died at age fifty-two of stomach cancer. Initially, Greg’s doctors did not consider the possibility that his disease was hereditary, but Greg suspected there was a link between his and his mother’s cancers. His sister, Karen, agreed. One day, while researching online, Karen came across a specific and very rare form of stomach cancer known as Hereditary Diffuse Gastric Cancer (HDGC). In 1998, scientists had found a mutation, in a gene called CDH1, which seemed to be largely responsible for five generations of early-onset HDGC in a Maori family in New Zealand. Several affected family members had developed gastric cancer as early as fourteen, with most succumbing to the disease in their forties. Since then, researchers have found more than one hundred families around the world who carry mutations in their CDH1 genes and, with those mutations, up to an 80 percent risk of developing stomach cancer. They have developed a genetic test to help predict if a patient is carrying a mutation and is likely to get (or has) HDGC.

Greg with daughter Johanna at the Relay for Life
Because the disease had been so recently identified, however, some of the doctors treating Greg in Wisconsin—in Stevens Point, Marshfield, and Madison—hadn’t even heard of HDGC. They concluded Greg had some form of gastric cancer of unknown origin, even though he had more than a decade to go before he reached age seventy, the average age of diagnosis for stomach cancers generally. And although doctors at the Houston cancer care center where Greg went for treatment were aware of HDGC, they didn’t advise Greg to get tested; after all, HDGC accounts for only 1 to 3 percent of gastric cancers. In 2007, medical centers tested for CDH1 mutations only if three family members—or two under the age of fifty—had been diagnosed with the disease. The Chelcun family didn’t fit that profile.
In fact, HDGC is a hereditary cancer that is almost unimaginably rare. As a result, statistics on global incidence are not well-characterized, But at a conservative estimate, if he were a randomly chosen person in the world’s population, Greg Chelcun’s chances of having a CDH1 mutation would be about one in nine million—actually, the odds were much better that Greg would become a millionaire by matching five of six numbers in the national Powerball lottery. Put another way, Greg Chelcun was far more likely to get struck by lightning (the odds of which are one in 10,000) than to have a CDH1 mutation.
But Greg and his sister, Karen, persisted. They had seen the beginnings of a pattern. They didn’t want to wait, as science and statistics seemed to demand, for a third young family member to be diagnosed with this terminal disease. After all, a disease that is rare in an entire population can feel less so when both you and your mother have suffered from it. Greg couldn’t bear the thought of not knowing whether he had unwittingly endangered his children. So he pushed hard to get tested.
It’s becoming increasingly important for patients to advocate for themselves in this way.
It’s becoming increasingly important for patients to advocate for themselves in this way. The strains and stresses on the American healthcare system are escalating at a furious pace, as healthcare providers and medical researchers try to balance the needs of more and more patients in the face of skyrocketing costs and dwindling resources and funds. Unsurprisingly, medical research priorities focus on understanding and developing treatments for widespread and high-profile diseases, like heart disease, diabetes, and the more common cancers. Similarly, medical education for doctors and health professionals focuses mainly on more common diseases, which is why some of Greg’s doctors did not even know about HDGC until Greg alerted them to its existence. Although more than 6,800 rare diseases are currently known and tracked by the National Institutes of Health’s Office of Rare Diseases Research (ORDR), in total they affect only about 9 percent of Americans. Finding clinical care providers who have experience diagnosing or treating such diseases can be a long, uphill battle for patients.
When doctors at MD Anderson Cancer Center in Houston, Texas, finally agreed to test Greg, they found that he did, in fact, carry a CDH1 mutation for HDGC. Greg and his family wouldn’t stop there; it was time to take medicine more actively into their own hands.
Chapter 3
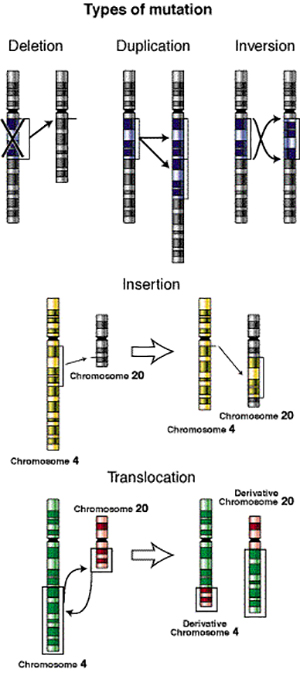
Types of genetic mutations
Mutations like the one Greg carried begin when a single letter in the three-billion-letter DNA sequence gets mutated accidentally to another DNA letter. A mutation may occur in just one of the millions of sperm cells that a man produces or in just one of the thousands of egg cell precursors that a woman produces. Much of the time, mutations occur in regions of the DNA that have very little effect on the proper functioning of the body, and anyway, relatively few eggs and sperm actually become people. In the case of the Chelcuns, two highly improbable things happened: in the reproductive organs of Greg’s mother’s mother (or mother’s father), an egg (or sperm) cell developed a single mutation in one copy of the CDH1 gene. Then, that egg or sperm cell became part of the embryo that became Greg’s mother. Through her, this mutated version of the gene entered the Chelcun gene pool, and with it, a 50 percent probability that each of Greg’s siblings, as well as Greg’s children, could draw the mutation.
Soon after getting his test results, Greg sat with his father, wife, son, daughter, and most of his extended family next to the teal waters of Lake Wisconsin. It was their annual reunion, and the air was thick with the scent of burgers and brats.
Unfortunately, Greg was about to deliver some solemn news that would dampen the festive mood, but he had to do it while everyone in the family was assembled together. He updated his family on the progression of his cancer and explained the genetic test he had taken. He tried to be upbeat and positive as he elaborated what the test result meant for many of those assembled. At that point, doctors weren’t sure when the mutation had first occurred in the Chelcun family. Had it begun in one of Greg’s grandparents? Or in one of his great-grandparents, or even farther back? Depending on where the mutation had arisen, all of Greg’s cousins and their children could be at risk for stomach cancer. The heavy import of Greg’s words settled in.
But Greg did have some “good” news: in a darkly ironic way, the Chelcuns were “lucky.” They could all get the genetic test, just as he had. And if they had the mutation, then there was a preventive measure they could take—highly drastic, but effective—that would prevent them from ever getting the cancer.
Greg had other news, as well. In order to help other families who might unwittingly be carrying CDH1 mutations, Greg’s sister, Karen Chelcun Schreiber, was establishing a community fund for stomach cancer research. (The fund, through Karen’s perseverance, would soon become a nonprofit foundation called “No Stomach for Cancer.”) Standing next to Greg, his wife, Cindy, held a basket of wristbands the same color as the lake. On them was the phrase Kia Kaha, which translates to “be strong in the face of challenge” in the language of New Zealand’s Maori people, where HDGC was first identified. The bracelets would go on sale for two dollars each, an initial fundraising drive.

Kia Kaha and No Stomach for Cancer wristbands. Image Courtesy of No Stomach for Cancer Foundation.
Also in the basket were mini-packs of tissues, placed there by Cindy, a psychotherapist and caretaker. After Greg had finished speaking, the family passed the basket around. Each of Greg’s relatives pulled out a bracelet, some also reaching for the tissues. The teal color of the bracelet had been chosen to represent the waters that connect all the people of the world. And soon the water was calling to them, too, and they were right back in it. Just like Greg, this family could not be kept down even by the most unlikely of probabilities.
Chapter 4
The National Institutes of Health (NIH) funds the majority of biomedical research in the United States and plays a large role in deciding which diseases to prioritize for developing therapies and treatments. To ensure that rare diseases are not ignored, the NIH founded the ORDR in 1993, then established the office by law when Congress passed the Rare Diseases Act in 2002. These efforts were meant to increase national investment in rare disease research at a time when the NIH budget was at an all-time high due to substantial increases in research funding that had been approved by Congress in the late 1990s. The past few years of fiscal restraint, however, have weighed heavily on the NIH. In the most recent sequestration cuts, the NIH budget was slashed by over $1 billion, bringing funding to below 2000 levels (in inflation-adjusted dollars) and severely affecting its ability to continue funding both common and rare disease research.
For their part, many biotech and pharmaceutical companies have shied away from developing treatments for rare diseases with small patient populations because the potential markets for any drugs they might develop are small. So, beginning with the Orphan Drug Act of 1983, both Congress and NIH have tried to incentivize research and development for so-called “orphan drugs” that target rare diseases. But even though more than two hundred orphan drugs have been approved and marketed in the United States in the past three decades since the Orphan Drug Act, the vast majority of rare diseases still have no viable cures or even treatments.
It may be up to grassroots foundations, like the one the Chelcuns started, to raise the money needed to fund research on debilitating or fatal rare diseases.
It may be up to grassroots foundations, like the one the Chelcuns started, to raise the money needed to fund research on debilitating or fatal rare diseases. Indeed, American patient advocacy groups have a successful history of such fundraising efforts, dating back to at least the 1950s. Only 15 percent of rare diseases have corresponding foundations that support research causes, but those that exist have successfully raised many millions of dollars for research. For example, two family foundations for Duchenne muscular dystrophy, the most common of rare, lethal childhood genetic disorders, have raised millions of dollars to support basic and clinical research, pushing treatments out of the lab and into human clinical trials. Hannah’s Hope Fund, cofounded by the parents of a child diagnosed with giant axonal neuropathy (GAN), a progressive nerve disorder with only 17 confirmed cases in the United States has raised nearly $3.2 million since its inception in 2008. These figures easily rival the funding dollars typically awarded by federal granting agencies to research on more common diseases.
Image Courtesy of Hannah’s Hope Fund
Research on rare diseases funded through patient and family advocacy foundations can have surprisingly far-reaching benefits. For example, research on rare diseases can inform research and treatment, as well as medical protocol, for other, more common diseases. The oldest rare disease advocacy organization in the United States is the National Tay-Sachs & Allied Diseases Association (NTSAD), founded by several affected families in 1957. It trailblazed new forms of community education around genetic diseases, built far-reaching family support programs, and successfully advocated for genetic screening services to be routinized in hospitals. Today, with a long history of driving scientific research, and its extensive patient and family network, it is recognized as a leader among nonprofit patient groups. NTSAD-funded research led to the discovery of the gene causing Tay-Sachs disease but, importantly, also led to the creation of carrier screening programs, which became the model for screening programs now used for hundreds of genetic diseases.
Rare diseases might affect only a few people, but rare disease research could have positive impacts for many.
The Chelcun family drew inspiration from these and other foundations in their efforts to build a patient advocacy group for HDGC. They realized that a foundation could fulfill many functions: bringing patients into communication with each other and with doctors, serving as an educational clearinghouse for important information on HDGC, and advocating for wider adoption of genetic testing for the disease. Rare diseases might affect only a few people, but rare disease research could have positive impacts for many.
Chapter 5
Johanna Chelcun, Greg’s daughter, who is now twenty-nine years old, recalls the last year of her father’s life vividly. Greg was an upbeat man, and even on chemotherapy, he rarely complained or got discouraged. He certainly never allowed his condition to take away his keen sense of humor. But those chemotherapies—docetaxel, 5-fluorouracil, and oxaliplatin, as well as some trial therapies, such as Avastin—had some severe and often frustrating side effects: numbness and tingling in his hands, which would cause him to drop and break plates or glasses, and sudden fevers and infections, which required frequent hospital visits. Greg’s body received trillions of molecules of anti-cancer drugs; they would kill some of Greg’s cancer cells, but the cancer proved too aggressive for the chemo to get them all.
Johanna has her mother’s auburn hair, her father’s apple-y cheeks, and her own green and brown-flecked eyes. The summer before her father died, she had blood drawn to find out whether she had inherited her father’s CDH1 gene mutation. Johanna had already had an initial meeting with a genetic counselor, who had advised her on the pros and cons of genetic testing. There was a one in two chance that Johanna had her family’s CDH1 mutation, as simple as a coin toss. If Johanna did have a CDH1 mutation, then she had an 80 percent probability of developing stomach cancer in her lifetime.

Johanna and Greg
As a budding physician assistant, Johanna Chelcun had seen patient blood draws a thousand times from the other side of the needle. The needle was sharp; the blood spurted out of it into the syringe, bubbly at first, then smoothly filling the vial. Embedded in that red, gushing liquid were millions of white blood cells, each one containing Johanna’s DNA, hundreds of meters of a thin white polymer, unraveling in the lab like a spool of thread, or a cobweb in the making. Somewhere among those billions of DNA bases in each cell were both copies of her CDH1 gene.
Chapter 6
Greg Chelcun and his sister, Karen, spent months during the last year of his life searching relentlessly for any information that might lead to a candidate disease explaining his symptoms. They finally learned about HDGC and the existence of the genetic test too late to save Greg’s life. But, in a curious reversal of roles in contemporary American medicine, Greg and Karen became medical authorities. They managed to teach Greg’s doctors about a disease many of them had never even heard of and to convince them that Greg needed the HDGC test.
Greg’s experience points to another crucial role that family foundations fulfill beyond raising money to fund research: they provide critical educational information not only to patients and their families, but also to their doctors. Karen remembers that when Greg was diagnosed, there was a basic lack of knowledge and support for stomach cancer families, leaving patients feeling isolated and alone. Now, through the website created by the Chelcun Family Fund, at least fifty new families with HDGC have been connected. The Foundation’s Facebook page followers have increased from seventy-six original members to more than 4,500 in May 2014.
Greg Chelcun in the hospital
Foundations connect patients who are so few they might otherwise never find each other, creating powerful communities of experts who have lived with the disease and know it better than the doctors who treat them. While it would be impossible for doctors to know everything about every rare disease, highly motivated patients can often research their conditions using online resources created and compiled by family foundations, greatly reducing their time to diagnosis. Patients also share information with each other about the latest treatments and therapies, including information about surgical procedures that can seriously impact their quality of life. Foundations also provide information about common treatment options, along with practical support, such as where to buy needed supplies. Karen Chelcun receives calls and email inquiries weekly from families newly affected by a stomach cancer diagnosis; increasingly, even genetic counselors are referring patients to the Chelcun Family Fund for the latest information about HDGC.
Rare diseases may turn out to be not so rare after all.
The Chelcuns hope that by raising awareness among patients, doctors, and hospital administrators, they can help change some of the rules that govern when and under what conditions genetic tests are offered. For example, they hope to encourage earlier HDGC genetic risk tests for other families, closer to the source mutation, rather than waiting for a family history of two or three generations to unfold. Though the criteria that patients must meet in order to be recommended by doctors for HDGC genetic risk testing have recently been updated, they are still too strict for the Chelcuns to have qualified even after Greg’s diagnosis: two or more cases of diffuse gastric cancer in a family, with at least one before age fifty. Both Greg and his mother got cancer in their fifties, so they wouldn’t have qualified for genetic testing under these criteria. Alternative criteria include three or more relatives diagnosed at any age; one relative diagnosed before age thirty-five; or one or more relatives diagnosed with diffuse gastric cancer and lobular breast cancer. Megan Harlan Fleischut, senior genetic counselor at Sloan-Kettering, suggests that criteria be made even more lax, listing several “red flags” that indicate a cancer might be hereditary: diagnosis of cancer at a significantly younger age than it ordinarily occurs; occurrence of the same cancer in more than one generation of a family; or occurrence of two or more cancers in the same patient or blood relatives. The Chelcun family would have met two of these criteria, and Greg might have learned he was at risk before he developed symptoms. Instead, as with most cases of stomach cancer, Greg’s doctors diagnosed the disease after the cancer had already grown and spread, too late to initiate effective treatment. More relaxed criteria would allow earlier testing for some families and almost certainly turn up many more cases of HDGC and CDH1 mutations, both in patients already suffering from HDGC and those at risk. Rare diseases may turn out to be not so rare after all.
Chapter 7
A month after her blood test, Johanna got into the car with her mother to drive to the clinic where she would receive the results. Cindy had flown from Wisconsin to Connecticut, where Johanna was attending college, to accompany her on this potentially difficult journey. So far, her aunt, Karen, had tested positive for the mutation. In different parts of the country, Johanna’s uncle, Ken, and her brother, Brian, would be receiving their results on this same day. I’m probably going to have it, Johanna thought.
Soon enough, she and her mother were at the hospital, sitting at a round table with her genetic counselor and an intern. Very calmly, the genetic counselor gave Johanna the verdict: “Well,” she said, “you tested positive.”
“I know,” Johanna responded. She knew this was an odd response, because she couldn’t possibly have known—the information hidden deep within her cells, an oddity on chromosome 16. But she’d had a feeling, so she wasn’t surprised. And she wasn’t particularly upset either—until her counselor began talking about how Johanna’s future might change given the information she had just received. If Johanna decided to have children, she would probably want to consider in vitro fertilization and genetic testing, selecting only the mutation-free embryos to bring to term. Johanna took solace in the possibility that she could stop the mutation that had caused so much suffering in her family. Yet a small, persistent thought formed in her head: If that had been an option for my mom and dad. . . .

Johanna at the hospital
Johanna’s uncle and her brother also tested positive. In fact, of the nine people in the Chelcun family who could potentially have the mutation, eight elected to get tested, and six tested positive. Each of those six now knew he or she had up to an 80 percent risk of developing HDGC.
Some of the older genetic tests—for example, tests for mutations causing Tay-Sachs disease, cystic fibrosis, or Huntington’s disease, which are all single-gene diseases—yield more certain results. In the case of Tay-Sachs, a disease where symptoms start at birth, there are viable treatment options; for others, like Huntington’s, which hits later in life, there are no known treatments, making positive test results feel like a death sentence.
Diagram of the stomach, showing
the different regions.
The test for the CDH1 mutation is more complicated. As most new genetics tests do, it predicts risk, rather than certainty, of disease. This is because the aberrant function of the mutated CDH1 gene is not the entire explanation for HDGC; some people who have the mutation appear not to have the disease. Researchers are still looking for other factors that contribute to one’s likelihood of getting this form of stomach cancer, but this uncertainty provides less than clear direction for patients.
The car ride back to Johanna’s apartment that day was silent. Johanna didn’t really want to talk about it although she was very calm and matter-of-fact about the whole thing, treating the news as one might treat a weather forecast for heavy rain. As with the decision to get tested, Johanna was not going to take any chances; although her probability of getting HDGC was 80 percent, there was a prevention option available to her that would guarantee she would not die from HDGC, as her grandmother had and her father soon would. But she would have to live the rest of her life without a stomach.
Chapter 8
Hacking off diseased tissue to save the patient is not a new idea.
Hysterectomies, mastectomies, limb amputations—hacking off diseased tissue to save the patient is not a new idea. Organ removal surgeries can be highly effective, but they are drastic and dramatically life-changing. Patients face difficult decisions about how much of their bodies they are willing to hack off in order to keep disease at bay. In a much-publicized recent case, celebrity Angelina Jolie opted to have both her breasts removed, based on her family history (her mother died of breast cancer at the age of fifty-six) and after testing positive for a genetic mutation that increased her likelihood of getting breast cancer to 87 percent. Those are high odds—a little higher than Johanna’s 80 percent likelihood of getting stomach cancer.

Johanna smiling post-surgery
Still, living stomach-less doesn’t at first seem possible. How can you live when there is nowhere to send the sustenance that keeps you alive? The stomach is a bit like a bowl of jelly, not quite set in its shape, changing when a person lies down or stands up. It can feel full, feel empty. When hungry, it can completely direct the activities of the body in which it is contained. Ancient physicians, such as the first century doctor Galen of Pergamon, believed the stomach alone, of all the body’s organs, had a mind of its own. Stomachs seem so animated, so closely entwined with emotions, that the prospect of living without one seems shocking. How could you feel butterflies when nervous or excited with no stomach to house the metaphorical creatures?
And yet, Johanna ultimately decided on a gastrectomy. After getting her positive genetic test result, she underwent cancer screening once a year, as advised by her doctors, for two years. Her biopsies all came back normal. But looking for cancerous cells on the vast inner surface of the stomach using random biopsies can be like looking for a needle in a haystack. So regardless of biopsy results, removal of the stomach is recommended before age thirty for anyone with the HDGC mutation and a family history of the disease.
“When I found out I had the mutation,” Johanna recalls, “I knew I was going to have the surgery. The question was just when.” Two years after her genetic test, on December 14, 2010, Johanna, then twenty-six years old, had her entire stomach surgically removed. (Johanna’s aunt, brother, and uncle also opted to go under the surgeon’s knife.)

Johanna and her fiance celebrate her “gastrectomy anniversary”
Removal of the slippery pinkish organ requires a two to three hour surgery, involving a series of complicated cuts and stitches, which totally remaps the geography of the digestive organs, repositioning the small bowel in relation to the esophagus. Johanna’s new “stomach” was made using her own body parts. Her surgeons cut a piece of her small bowel, pulled it up, and sewed it into the shape of a small pouch about the size of a soda can.
The pouch is not really a stomach, though; it holds food but cannot digest it. When Johanna swallows, food and drink move from her esophagus through this pouch directly into her small bowel. Digestion takes place there, aided by digestive juices from her pancreas and common bile duct, which enter through the duodenum. Without a stomach, Johanna no longer feels hunger, and there are certain things—milk, yogurt, too much sugar—she simply cannot eat because she cannot digest them. She must eat in very small portions throughout the day. Overeating can put her on the couch, in great pain. She receives regular vitamin shots, and both she and her doctors closely monitor her weight and nutrition.
But in addition to her father’s smile and CDH1 mutation, Johanna also has his optimism: as far as eating goes, she rates her life sans stomach an 8.5 out of ten. And every year, on December 14, she and her fiancé go out for a small meal to celebrate her “gastrectomy anniversary.”
Chapter 9
Cindy Chelcun once remarked of her late husband, Greg, that he understood life in complex ways that were often only conveyed on the stage. Stages played an important part in Greg’s life as an educator: he studied dance and performed onstage at the University of Illinois in the late ’60s and ’70s, then nurtured that talent in his students at Stevens Point Area Senior High, where he produced, directed, and choreographed many successful musicals.

Johanna’s uncle Ken, aunt Karen, and brother, Brian, show off their surgical scars
And so it was fitting that, on July 18, 2009, a celebration of Greg’s life was held on the very stage where he had not only persuaded many teachers over the years to contribute scholarship money to students, but also directed those beloved musicals. Even on that day, the stage took on multiple roles—theatrical, emotional, educational, philanthropic—in Greg’s memory. Four hundred people—friends, family members, and former students—filed into the auditorium for this celebration of Greg’s life. The stage was hung with colorful backdrops from all the musicals he had directed: Cats, Les Misérables, Evita, and more. Group after group of students and friends played, sang, and performed. But they were also introduced to the newly established Chelcun Family Fund for Stomach Cancer Research and its mission: to expedite education and research for screening, early diagnosis, treatment, and prevention of HDGC. Many attendees bought Kia Kaha bracelets and donated money to the cause.
The more the Chelcuns bond with other families who share the trials and tribulations of HDGC, the less rare and improbable their own condition seems to be.
Since that day, the fund has blossomed into a formal research foundation. No Stomach for Cancer is a 501(c)(3) nonprofit organization that supports research, educating the public and medical professionals, and uniting and empowering people worldwide affected by stomach cancer. The foundation’s website, originally created and managed by Greg’s sister, Karen, has drawn hits from people in more than thirty countries, with over 129,000 unique visits. After two years and much perseverance, Cindy finally managed to convince the local healthcare center, where Greg first went with his symptoms, to do a formal physician education program on HDGC. Brian, Johanna’s brother, who worked on Capitol Hill, helped the family navigate the legalities of officially making November “Stomach Cancer Awareness Month.” And all of the Chelcuns are gaining recognition within the medical community as HDGC experts. Johanna gave a guest lecture for her graduate-level genetics class at Quinnipiac University, where she now works and teaches, and was interviewed about HDGC at Yale. In November 2012, the foundation sponsored the 1st Annual No Stomach For Cancer Walk; participants from thirty-five states and ten countries raised over $35,000 for stomach cancer research. In all, No Stomach for Cancer has raised more than $150,000 through donations and grants to support research into possible treatments for the disease. The foundation has also connected many people around the world affected by the disease, as well as their supporters and well-wishers. Although only one hundred or so families in the world have been reported in the medical literature as being affected by HDGC, Greg’s wife, Cindy, is convinced the actual number is much higher. Karen believes it may be as high as 150. The more the Chelcuns bond with other families who share the trials and tribulations of HDGC, the less rare and improbable their own condition seems to be.
Chapter 10
The Chelcun Family Fund for Cancer Research has blossomed into a formal research foundation, “No Stomach for Cancer.” Image Courtesy of the No Stomach for Cancer Foundation.
There are few certainties when it comes to diseases as complex as stomach cancer, and elective surgeries, odds or no odds, are not for everyone. No Stomach for Cancer is hard at work building networks through which affected individuals can share their stories and experiences, and offer solidarity, comfort, and solace to others dealing with the difficulties of adjusting to life in a surgically reworked body. But the Chelcuns also hope their foundation can raise enough money and awareness to make HDGC a higher research priority in the medical community, by offering grants to medical researchers developing less invasive treatments, for example, so that gastrectomy isn’t the only option for those with a CDH1 mutation.
Investing time and money in rare disease research can seem like an inefficient use of resources, but it is infinitely important to the people, however few they may be, who benefit. Until 1998, HDGC didn’t even “exist” as a subtype, and doctors treated it as they would any other stomach cancer. Indeed, had researchers continued to treat stomach cancer as a singular disease with a singular cause rather than looking for rarer forms of the disease, they would never have come across the HDGC in families around the world and would never have devised a genetic test. Families like the Chelcuns would have paid the price, with devastating consequences. In this way, policy debates about what kinds of research to fund are not just matters of economics; they also involve ethical questions about people’s values, their stakes and priorities, and about their futures. Johanna, her brother, her aunt, and other Chelcuns who have defeated HDGC represent one return on that investment, and patient advocacy groups like No Stomach for Cancer are important stakeholders in the decision-making processes that guide biomedical research.

2012 No Stomach for Cancer Walk. Image courtesy No Stomach for Cancer Foundation.
HDGC may seem rare today, but rare and common are relative terms. For the Chelcuns, they are terms that have become less meaningful as, every year, more and more families have discovered they’re affected with CDH1 mutations. And as medical researchers continue to identify more rare diseases that can be predicted with relatively high certainty, we may all discover ourselves in a camp of a few hundred individuals, for some disease or other. Common isn’t necessarily common, and rare isn’t necessarily rare. In such a light, funding priorities take on an entirely different sheen.
As did many of Greg’s favorite songs, when sung by his former students at the memorial. When Greg was alive, he would often break into “The Banana Boat Song” at both predictable and unpredictable times, to turn a tense moment into a light one. He did it to make people laugh—both with him and at him. On the day of his memorial, several of Greg’s former students sang the song in his honor, the upbeat refrain, “Daylight come and me wan’ go home,” filled with new resonance. In more ways than one, Greg Chelcun had clearly left an important and lasting legacy: in the voices of his former students as they sang and performed, and in the voices of the family and foundation he left behind.

